�l(f��)���r�g��2020-03-21���ٷ���Ƽ�Փ���g�[��1��
ժ Ҫ�� ժҪ��Ŀ��̽����ˎƷ����Ʒ�˻���ɫ�V�l����Ӱ����ء�������2�����˻��Č���Ʒ����������HPLCDAD�����伃���M���A(y��)�˻����Y(ji��)����HPLC��ɫ�V����͡�������ı��������ء����١��z�y���L���伃�șz�����һ����Ӱ푡��Y(ji��)Փɫ�V�l����Ӱ������^�࣬�ڌ��H��(y��ng)��
����ժҪ��Ŀ��̽����ˎƷ����Ʒ�˻���ɫ�V�l����Ӱ����ء�������2�����˻��Č���Ʒ����������HPLC—DAD�����伃���M���A(y��)�˻����Y(ji��)����HPLC��ɫ�V����͡�������ı��������ء����١��z�y���L���伃�șz�����һ����Ӱ푡��Y(ji��)Փɫ�V�l����Ӱ������^�࣬�ڌ��H��(y��ng)���пɾC�Ͽ��]��
�����P(gu��n)�I�~�����ȷ���;����Ʒ;��ЧҺ��ɫ�Vһ��������Йz�y
�������W(xu��)����Ʒ���ڽY(ji��)��(g��u)�����|(zh��)��ȫ����Ļ��A(ch��)�ϣ����������ͻ��W(xu��)�yԇ�r�����c��ԇˎƷ�M�Ќ��յ����|(zh��)���伃���m����ʹ��Ҫ�����|(zh��)����һ�Č����Ʒ[1]�����҇�������Ʒ�ľ��Ƽ��˻����b�����Ї�ʳƷˎƷ�z���о�Ժ(ԭ�Ї�ˎƷ������Ʒ�z���������Q�Йz��)�Г���2010�꣬�������c�˲����°�ˎ���û��W(xu��)����Ʒ�Ę˻��������������Ї�ˎ�䡷2010��挦���ˌ���Ʒ�M�м��ȷ����⣬߀���c����ˎ��(USP)���W��ˎ��(EP)��Ӣ��ˎ��(BP)���ձ�ˎ��(JP)�ȷ����Y(ji��)�����^����������ˎ�䷽���ĽY(ji��)����ԡ�������2������Ʒ������̽ӑ�ڌ�����Ʒ�M�м��șz��rɫ�V�l����Ӱ����ء�
����1�x���cԇˎ
����Waters2690—996���O����Йz�y������ЧҺ��ɫ�Vϵ�y(t��ng)(Waters��˾)��AE-240�����ƽ(÷���չ�˾)��ZFһ1������������x(�Ϻ��늹�x���S)����������������Ʒ(��̖��100859)���h(hu��n)����������Ʒ(��̖��101118��201001)(�Ї�ʳƷˎƷ�z���о�Ժ)���״����������ɫ�V��������ԇ�������������ˮ�鼃��ˮ��
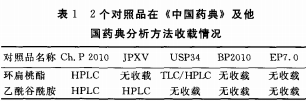
����2���Ї�ˎ�估��������ˎ�����d����������r
������2������Ʒ�ڡ��Ї�ˎ�䡷����������ˎ�����d��r�M�Йz���������������d��rҊ��1��
����3����Ʒ�˻��Ќ�ɫ�V��ļ���Ҫ��
��������Ʒ�˶����I(y��)ָ��(d��o)�����_ָ�����M�����á��Ї�ˎ�䡷��Һ����ߚ�����M�м��șz�飬�������ˎ���ɫ�Vϵ�y(t��ng)�c���Ї�ˎ�䡷��һ�£�Ҳ��ͬ�r���졣HPLC������PDA�z�y����������e�wһ���������ɷ����팦�շ�����ԇƷ��ȵĴ_������0.1�������ȴ���10����A(ch��)������ԇƷ��Һ�������M�м��șz�飬����������ɫ�V�l���M���{(di��o)��ֱ�����șz��ͨ�^;��Ҋɫ�V�l���ă�(y��u)���̶�ֱ���P(gu��n)ϵ���弃�ȡ�
����“�弃��”�yԇ�nj�ij�����еĹ��V�c�����c���V�M�б��^���Դ_��ԓ���Ƿ�������V��ͬ�ĽM��_2]��PDA���ԫ@�Ø�Ʒ��ɫ�V�D��ÿ��ɫ�V�M�ֵĹ��V�D�����ṩ���S���Vһɫ�V�D(3D-�V�D)�������c��������Waters2690—996�����R�eҊ�D1�������c������ļ��ȽǶȼ������ֵ�Y(ji��)��Ҋ��2��
����4ԇ�
����4.1����������HPIC���Ș˻��^�̵�Ӱ�����
����4.1.1ɫ�V�����x��
�����ڌ���������������Ʒ�˶��У�����3�N��ͬ�S�ҵ�ɫ�V����LichrospherC18��(4.6mm×250mm��5/~m)(�h��Ƽ�);XBridge��C18��(4.6ramx250mm��5gin)(�V�ݷ��_�T);CapcellPAKC18��(4.6mm×250mm��5/~m)(�ձ��Y����)�������ࣺ0.05mol·I2����������Һ(��1o�����{(di��o)��(ji��)pHֵ��2.99)һ�״�(95��5)41;�z�y���L��210nm;�M���w�e��2OL;���٣�1.0mL·min;���أ�3O��;DAD���貨�L��190��800nm;��ԇƷ��Һ�ĝ�ȣ�lmg·mI;����Ʒ��Һ�ĝ�ȣ�1p.g·mI���������������ڲ�ͬ�S��ɫ�V����ɫ�V�DҊ�D2��
�����ĈD2��ҊLichrospherC18ɫ�V��(4.6ram×250mm��5/~m)(�h��Ƽ�)�����^�ã�������δͨ�^(���ȽǶȞ�4.532�������ֵ��0.349)������x��LichrospherC18����ԓƷ�N���Ȝyԇ��ɫ�V����
����4.1.2����������ĵ�(ji��)
����ɫ�V��ı����r�g�^�̣����������m���{(di��o)��(ji��)���l(f��)�F(xi��n)���ЙC���{(di��o)��(ji��)��1�r��ɫ�V��ı����r�g��5min���ң��f�������������匦�״������жȲ��ߣ��ҷ弃��δͨ�^�����ԏ����ء����١��M�����M���m�����{(di��o)��(ji��)��
����4.1.3DAD���貨�L���x��
�������貨�L�r�l(f��)�F(xi��n)�״��Ļ��|(zh��)Ӱ��^���������������״��Ľ�ֹ���L��205nm�����O(sh��)��DADɫ�V�弃�ȽǶȾ��Ǽ����ֵ��10�����ϣ�����δͨ�^��������ԓӰ푣��P�ߌ�DAD���貨�L�O(sh��)����205��800nm��
����4.1.4���ء����١��M�������x��
������ԇ��У��������m�����ͣ����ٽ��ͣ�ɫ�V�屣���r�g��llmin���ң��弃�ȽǶȞ�0.498���������ֵ��0.358������δͨ�^;�ڱ��C0.1������Һ���������Ȟ�17.3�r�����M�����{(di��o)��(ji��)��1oL������������ɫ�V�弃�ȽǶȞ�0.268�������ֵ��0.308������ͨ�^��
������Ҋ����ʹɫ�V�弃��ͨ�^���M��0.1������Һ��ɫ�V�������Ȟ�1O�Ļ��A(ch��)�ϣ�ɫ�V����͡����貨�L������������M�ɳɷֱ�����׃�������ء����١��M�������x��ɫ�V��ļ�����һ����Ӱ푡�
����4.2�h(hu��n)���������P(gu��n)���|(zh��)�z�y���L���x��
�����ڌ���Ʒ�˻��У����Ȝy�������x��ɫ�V������HPLC�z����ö��O����Йz�y���z�yɫ�V�弃�ȣ�TLC�z��Y(ji��)��������HPLC�z��Y(ji��)���ą�������
�����h(hu��n)����������Ʒ�˻�HPLC���ȷ���ɫ�V�l������CapcellPAKC18(4.6mm×250ram��5m)��ɫ�V��������һˮ(4��1)�������࣬�z�y���L��228nm(�����cUSP32һNF27��ͬ)��HPLC���șz�y�Y(ji��)��������e�wһ��������ȡ228nm��ɫ�V�D���õĹ�ԇƷ��Һ�п��s�|(zh��)������0.20(���˙z(sh��)��(j��)�ı����ԣ��o���Ĕ�(sh��)ֵ�Dž���ֵ)����������s�|(zh��)�ĺ�����0.2(���˙z(sh��)��(j��)�ı����ԣ��o���Ĕ�(sh��)ֵ�Dž���ֵ)��
�����h(hu��n)������TLC���z�鼃�ȱ���ɫ�V�l�������zGF����鱡�Ӱ壬������һ��������һ������(8��2��1��v/V)��չ�_������������(254nm)�zҕ[6]��TIc���șz�y�Y(ji��)����ԇƷ�@1���s�|(zh��)���c�����С���ɫ�c4������Һ�ஔ(���˙z(sh��)��(j��)�ı����ԣ��o���Ĕ�(sh��)ֵ�Dž���ֵ)��
�����s�|(zh��)�z�y�Y(ji��)�����ò�ͬ��ɫ�V�������Y(ji��)������^����ԭ����Ҫ���s�|(zh��)�ęz�y���L���x��(d��o)�½Y(ji��)���IJ�ԡ���HPLC�����У����z�y���L�O(sh��)����254nm��������e�wһ��������ԇƷ��Һ�п��s�|(zh��)������4.34(���˙z(sh��)��(j��)�ı����ԣ��o���Ĕ�(sh��)ֵ�Dž���ֵ)����������s�|(zh��)�ĺ�����4.1%(���˙z(sh��)��(j��)�ı����ԣ��o���Ĕ�(sh��)ֵ�Dž���ֵ)����ԓ�s�|(zh��)��254nm�IJ��L̎��������ա��Y(ji��)���cTLC�z�y�Y(ji��)�����\���ϡ��P����ȡ228nm���L̎�h(hu��n)���������s�|(zh��)��������r������(j��)��푑�(y��ng)���ӣ��_��228nm��h(hu��n)�����������P(gu��n)���|(zh��)�ęz�y���L����Ҋ�z�y���L���x����HPLC���P(gu��n)���|(zh��)�z�y�е���Ҫ�ԡ�
�������]��x����Ո���W(xu��)���̎���Փ����ôͶ��
����5ӑՓ
�����P���ڌ�����������HPLC���Ș˻�ԇ��аl(f��)�F(xi��n)�����ȣ�ɫ�V������͡����ϵ����|(zh��)����β��r��ɫ�V��ķ����кܴ��Ӱ�;��Σ����]���܄��Ľ�ֹ���L���{(di��o)��(ji��)DAD���貨�L����;�ٴΣ����]������ĽM�ɳɷֱ�����׃�������ء����١��M�������x��Ҋ��ɫ�V��ļ��șz�y��Ӱ������^�࣬���hҪ�C�Ͽ��]�@ЩӰ����ء�
������HPLC���M�к����y������TLC�b�e�r������߷������`���ȣ����ɔ_�������x�����ɷֵ�������ղ��L����z�y���L���������P(gu��n)���|(zh��)�z��Č������s�|(zh��)������ˎ��������ղ��L�_����z�y���L���t�s�|(zh��)�ڴ˲��L�µ����տ���ƫ�ͣ�ijЩ�s�|(zh��)�����o���գ��@�ӕ���Ɍ��s�|(zh��)�����ĵ�����©�z���Ķ����ܷ�ӳ�a(ch��n)Ʒ���挍�|(zh��)����Ӱ��ˌ�Ʒ�N�|(zh��)���ɿ��Լ���(w��n)���Ե��u�r��