�l���r�g��2021-09-27���ٷ�����̎��QՓ���g�[��1��
ժ Ҫ�� ժҪ��Ŀ�Ľ���HPLC���y���}�ᰱ�����ڷ�Һ�����Ǻ�ɽ�洼�����ķ�����������ȫ��������20�����a��I��������r����������HYPERSIL����ɫ�V��(4.6mm250mm��5m)��������-ˮ(80�s20��V/V)�������࣬����1.0mLmin-1�����؞�35�棬ʾ���۹�z�y�����Y���}�ᰱ��
����ժҪ��Ŀ�Ľ���HPLC���y���}�ᰱ�����ڷ�Һ�����Ǻ�ɽ�洼�����ķ�����������ȫ��������20�����a��I��������r����������HYPERSIL����ɫ�V��(4.6mm×250mm��5μm)��������-ˮ(80�s20��V/V)�������࣬����1.0mL·min-1�����؞�35�棬ʾ���۹�z�y�����Y���}�ᰱ�����c���Ǻ�ɽ�洼�������÷��x�����քe��1~10mg·mL-1�����ȣ�����(r=0.9996��n=6)��ɽ�洼(r=0.9999��n=6)�����Pϵ����;�����քe��8.03��8.04μg;ƽ��������(n=9)�քe��101.0%��100.9%��RSD�քe��1.03%��0.97%����ȫ��������20�����a��I�Ę�Ʒ�M�Мy�������e��I�y���ĽY���c���a̎����һ�¡��YՓ���������������У��ʴ_�ɿ����؏������ã���y���ڷ���Һ�гCζ����������r���|���u�r�춨�˻��A��
�����P�I�~���}�ᰱ�����ڷ�Һ;����;ɽ�洼;��ЧҺ��ɫ�V;ʾ���۹�
�����ڷ���Һ���������ӳCζ���������wˎ����̡����Ϳ�ζ��������Ȼ���˹��ϳɶ��������ζ�����˹��ϳɵģ������z����a��һ���Ķ������ã������¸�Ѫ�ǡ����ֵȴ��x�Լ���������߀��һ�����°���[1-2]���҃�ͯ��ڷ���Һ����Ҫʹ��Ⱥ�w��������ζ����ʹ���^���б�횇����������[3]��
�����˴·��u���ռ���22�����a��I���}�ᰱ�����ڷ�Һ���������a�S���ṩ�����a��ˇ���Y�Ϸ������ڷ���Һ��ʹ����ζ����Ҫ��˾��̹��AK�ǡ��Ǿ��c��ɽ�洼�����ǵȡ�����īI�����Ј������������z�y�ķ����������ڛ]���������յ����ǡ�ɽ�洼��δ�ܙz�y[4-10]�����˃ɷN�Cζ��������ࡣ���Ľ����˸�ЧҺ��ɫ�V-ʾ���۹�z�y�ķ����y�����ǡ�ɽ�洼����������ͬ�r������ȫ��������ԓ�aƷ��������r����˾��̹��AK�ǡ��Ǿ��c�ͱ��aƷ���־������ø�ЧҺ��ɫ�V-����z�y���z�y(�����о����)��
����1�x���cԇ��
����1.1�x��Agilent1260��ЧҺ��ɫ�V�x(���DAD�z�y��)����ʿ÷������ӷ�����ƽ(��̖��XPE205)����ˮ�C(����Millipore��˾)��
����1.2ԇˎ��ԇ��
����ԇˎ��22�����a��I��63����Ʒ������2020������u�r�Գ���Ʒ��ɽ�洼(��̖��F675G����I�ṩ);����(��̖��FL1001200311-1����I�ṩ)��ԇ��������(Sigma��ɫ�V��)������ˮ��
����2�����c�Y��
����2.1ɫ�V�l��
��������HYPERSIL������(4.6mm×250mm��5μm);�����ࣺ����-ˮ(80�s20��V/V);ʾ���۹�z�y��;���أ�35��;���٣�1.0mL·min-1;�M���w�e��20µL��
����2.2��Һ���Ƃ�
����2.2.1ϵ�y�m������Һ�քeȡɽ�洼������ԭ�ϸ��m������ˮ�ܽⲢϡ��Ƴ�5mg·mL-1����Һ������ϵ�y�m������Һ��ϵ�y�m����ɫ�V�DҊ�D1����Փ�唵�քe��ɽ�洼������Ӌ�㣬������2000��ɽ�洼�c���ǵķ��x�ȑ���С��1.5��
����2.2.2�˜��������Ƃ�քeȡɽ�洼������ԭ�ϸ��m������ˮ�ܽⲢϡ��Ƴ�10mg·mL-1����Һ���錦��Ʒ�A����Һ��������Ʒ�A����Һ��ϡጳɝ�ȷքe��1��3��4��6��10mg·mL-1�ľ��Ԍ�����Һ��
����2.2.3��ԇƷ��Һ���Ƃ����ɽ�洼�����ǵ�̎������������ȡ�}�ᰱ�����ڷ���Һ�m������ˮ�ܽⲢϡ��Ƴɸ���ɽ�洼�����Ƿքe��3~6mg·mL-1����Һ���u�����鹩ԇƷ��Һ��
����2.3�����W��C
����2.3.1�����Pϵ���������a��I̎���漰��ζ���ľ��Է�����4�N��������ɽ�洼�����ǘ˜�������Һ���÷�������“2.2.2”헡��Ԍ�����ɫ�V�D����e��v����(y)���|����Ȟ�M����(x��mg·mL-1)�M�лؚw���������Է��̷քe�飺ɽ�洼y=2.15×105x-7.45×104��r=0.9996;����y=2.15×105x-1.51×105��r=0.9999���Y��������ɽ�洼�������ڸ��ԝ�ȷ����Ⱦ����Pϵ���á�
�������P֪�R���]�����^���װl��ˎ�W�Փ�ĵ�ʡ�������ڿ�??
����2.3.2�؏���ԇ���I18�����a̎����������������������ɽ�洼�����x����I18��ijһ����Ʒ(��̖��200107)����“2.2.3”���ƽ���Ƃ�6����Һ���M���؏��Կ��죬6�ݘ�Ʒ��ɽ�洼�����ǜy�ú�����RSD���քe��0.62%��1.44%����С��2.0%��
����2.3.3���ܶ�ԇ������Һ(6mg·mL-1)�؏��M��6�Σ�ɽ�洼�����Ƿ���e��RSD�քe��1.75%��1.70%����С��2.0%��
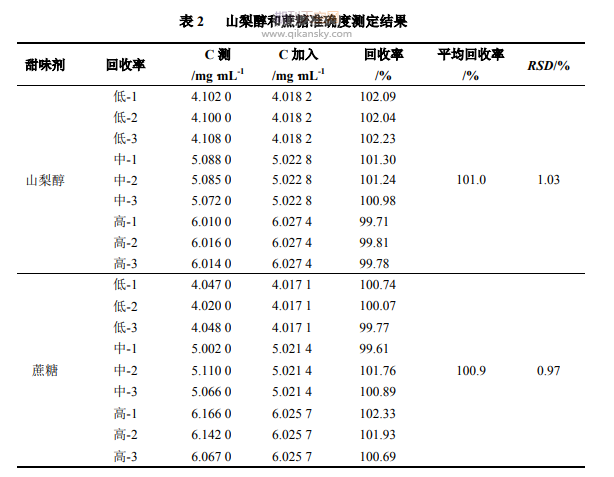
����2.3.4�ʴ_��ԇ���^ɽ�洼�������⣬̎���е������M�ְ���̎�����������հ��|���ٌ�ɽ�洼�����nj����A��Һ����̎��80%��100%��120%���룬ÿ�����ƽ���Ƃ�3�ݘ�Ʒ����9�ݘ�Ʒ�����չ�ԇƷ�Ƃ䷽��ͬ��̎�������á��y���Y��Ҋ��2��
��������ɽ�洼��̎���еĺ�����5%~30%֮�g��������̎���еĺ�����0.1%~30%֮�g������2020����Ї�ˎ���IJ�9101Ҏ��[11]��ɽ�洼�����ǵ�ƽ�������ʷքe��101.1%��100.9%��RSD�քe��1.03%��0.97%��������Ҏ���������������ķ��y��2�N��ζ�������ʴ_�����á�
����2.3.5�z�y�Ͷ������Ծ���1mg·mL-1��Һ��ϡጣ���S/N=3�ĝ�Ȟ�z�y�ޣ���S/N=10�ĝ�Ȟ鶨���ޣ��Y��Ҋ��3��
����2.3.6������ԇ�ȡ�հ���Һ�������ࡢ��ɽ�洼��������o�ϻ����Һ���}�ᰱ������Һ�M�ӣ���ɽ�洼������ɫ�V��λ��̎��δ���壬�C���o�ɔ_��ɫ�V�DҊ�D2��
����2.4��Ʒ�y�������������ķ�����20�����a�S��50����Ʒ�M�Мy����������˷��������Y��Ҋ��4����Ʒ���P�D�VҊ�D3��
���������a��I��Ʒɽ�洼�c���ǵĜy�������c�����a̎�����^�����ϱ���֪��ɽ�洼�Ĝy���Y��ռ̎������Ȟ�94%~107%���c̎������һ��;�����ǵĜy���Y��ռ̎������Ȟ�19%~100%��������I�y���Y���^������ƫ�ͣ��c̎��Ҏ�������^����롣
����3ӑՓ
����3.1ɫ�V�����x��
��������_ʼ�x���Y����CAPCELLPAKC18MG��(4.6mm×250mm��5μm)ɫ�V������������-ˮ�wϵ����׃������ɽ�洼�����Ǿ�δ���塣���Q��ThermcAPS-2HYPERSIL������(4.6mm×250mm��5μm)�������������������-ˮ�wϵ���z��ɽ�洼�����ǣ��������÷��x�ȡ�
����3.2��������x��
�����x�ü״�-���_�}�wϵ��ɫ�V�����^��o�����_���y�ɷ֡����Q������-ˮ�wϵ�����εõ��ܴ���ƣ��������Խ���x��׃�ã���K�xȡ����-ˮ(80�s20��V/V)���������ࡣ
����3.3�y���Y������
�������Ό�����y��������ռ̎������Ȟ�19%~100%��������I�y���Y���^������ƫ���^�࣬�c̎��Ҏ�������^����롣����ԭ��һ�dzCζ�����벻��;���������a�^�����Гpʧ���Cζ���Pϵ���H�Pϵ���aƷ�ĿڸУ����Pϵ�������|����������A�ȡ�����ָ�˵ȡ����h��I����ԭ����Ҏ���Cζ��ʹ�á�——Փ�����ߣ���С����ϯ־���������A����������������
SCISSCIAHCI