�l���r�g��2020-04-29���ٷ���r�IՓ���g�[��1��
ժ Ҫ�� ժҪ�|�ӽ��QĤȼ��늳��������;Gɫ��Դ���g����δ��늄���܇����ɢʽ�վ���I���зdz��V韵đ���ǰ������Ŀǰ�Ƽsȼ��늳��̘I������Ҫƿ�i����߳ɱ��͵͉��������F������߀ԭ������ʹ���dzɱ�����Ҫ��Դ��Ҳ�ǛQ��ȼ��늳����ܵ��P�I���ء����ľC��
����ժҪ�|�ӽ��QĤȼ��늳��������;Gɫ��Դ���g����δ��늄���܇����ɢʽ�վ���I���зdz��V韵đ���ǰ������Ŀǰ�Ƽsȼ��늳��̘I������Ҫƿ�i����߳ɱ��͵͉��������F������߀ԭ������ʹ���dzɱ�����Ҫ��Դ��Ҳ�ǛQ��ȼ��늳����ܵ��P�I���ء����ľC����һ�N���͵���߀ԭ����——�K�������g�������B��Y�������Լ��^�ɽ��ٽM�Ɍ���߀ԭ���������Ժͷ����Ե�Ӱ푣�����K�������İlչ�o����չ����
�����P�I�~�|�ӽ��QĤȼ��늳أ���߀ԭ������늴����������g������
����1.����
������ȫ����ԴΣ�C�Լ��h�����}���������ć����΄��£��lչ�坍�c��������Դ�ѳɞ�������������P�ĵ���Ҫ�h�}���ڸ�ɳ��m�lչ������Դ�У���������г�ɫ�ı������ܶȃ��ݼ��V���ļ��g�m���ԣ����J����δ������аlչǰ��������Դ֮һ���|�ӽ��QĤȼ��늳�(ProtonExchangeMembraneFuelCells,PEMFCs)���g����������еĻ��W��ֱ���D������ܣ��ǚ���������ֱ����Ч�ķ�ʽ[1]������ʮ�꣬PEMFCs���g�ѵõ�Ѹ�ٰlչ��Ŀǰ���V���đ��þ�������늄���܇����Դ��2014���ձ��S�﹫˾���a���̘I����ȼ�����܇(FuelCellVehicle,FCV)Mirai��־��ȼ��늳��̘I���r���ĵ���[2]��
������ȼ��늳،��H�����У�ꎘO��߀ԭ����(OxygenReductionReaction,ORR)��һ�������W���������䷴������ֱ�ӛQ����늳ص����ܣ���Ҫʹ���F����Pt�������[3]��Ȼ��Pt��ϡ�н��٣�ȫ�������ޣ��r���F����ʹĿǰPEMFCs���R�ɱ��ߵĆ��}����һ���棬Ŀǰ�V��ʹ�õ�Pt/C�������L�r�g�����l���´��ڼ{���w���F�ۡ��ܽ⡢�ٳ��e�Լ�Ó��Ć��}��ʹ���������@���½���늳ط������ܲ��á����˽��ʹ�����Pt��������������������о����c��Pt�c�^�ɽ���M(M=Fe,Co,Ni,Cu,Pd��)�γɵĺϽ������Ŀǰ�����S��Ͻ���������P�;C��������[3][4][5]������F��Ͱlչ����o�����uՓ�����ǣ���ȼ��늳�ꎘO������늉������ԗl���£��Ͻ�����е��^�ɽ��ٴ������ܳ�������Ⱦ�|�ӽ��QĤ�Ć��}��ʹ늳�����Ѹ��˥�p���������һ�N���͵�ORR����——��������Y���Ľ����g�����������F�����ߵĴ����Ժͷ����ԣ������ˇ������о������ߵďV���Pע�����Č�������Щ�ꌦ���@������о����Ĵ����ĽM�ɡ��Y�������������Ƕȳ��l���C������͵Ľ����g������ORR�������о��Mչ�����������R������Ͱlչ����o��һЩչ����
����2.�����g�����������Y���c������
�������ڼ��K�������о���ORR�����C���ܶȷ�����Փ(DensityFunctionalTheory,DFT)Ӌ���������Ч��߀ԭ�����������Ӡ�B����������������ӵ���������ʹO=O�p�I���ѣ��������ڷ������g�a��Ĵ�������Ó�x��ʹ��������M�С�Nørskov����[6]��Ӌ���@ʾ���������K���挦�������ӵĽY�����^�ڏ��������K�����M�к����{�أ�ʹ����挦����������ܱ�Pt{111}�������s0.2eV�������������ԡ������@�ӵ���Փ��PtM�Ͻ������������V���о����䌦��ORR�����Ե������Ҫ���F�ڃɂ����棺��Ч�������Ч����һ���棬�����^�ɽ���ԭ��ֱ�����Kԭ��С���Ͻ�����ʹԭ�е��K-�Kԭ���g���տs(��Ч��);��һ���棬�����^�ɽ��ٸ�����ʧ��ӣ�ʹ�Ͻ��w��������ӽY���l����׃(���Ч��)�����ߵąfͬ��������������������w��������pַ�������Լ������a���Ó�����Ķ��Mһ����ߴ�����[7]�����ǣ����@Ͻ��У��^�ɽ���ԭ���Թ����w��ʽ�o��ē��s���K�����У�ԭ�������Ա���ԭ���K�����������Y��(face-centeredcubic,fcc)���Ͻ�������a���Ď�Ч�������Ч������������������Ե����Ҳ���ޡ�ͬ�r���o����^�ɽ���ԭ������ʹ�������ԗl���º����ĺϽ��w�����ܳ��������L�r�gѭ�h�^�����^�ɽ����ܽ�Ć��}��ʹ������Ѹ���½���
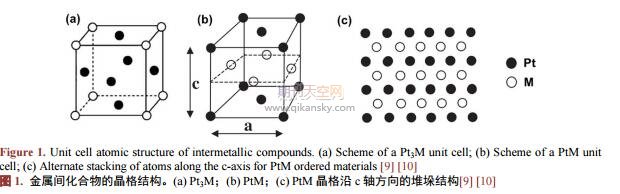
���������g���������ɃɷN���N���ٽMԪ�������Y���γɾ��в�ͬ�������M��Ԫ�ص��L�������w�Y�������Ҿ��н��ٻ������ԵĻ�����c�Ͻ�Y����ͬ���ڽ����g������ľ���Y���У�Pt��Mԭ�Ӷ������ռ���������������c�����Խ����I���x���I����ã�ʹ�������w�ʬF���L������ľ�ϵ�Y�����@�N�L������ĽY��ʹ���������Ĵ�����[8]��������ͬԭ�ӱ�����һ������g������֞�Pt3M��PtM��PtM3�࣬��D1��ʾ����Pt3M��(�D1(a))��Ptԭ��ռ�������w���������������λ�ã���Mԭ��ռ�������w�İ˂���c(PtM3�෴);����PtM�У�Pt�cM��ԭ�ӌӽ�������(�D1(b)~(c))���@�Nԭ�����о��������ķ��Y��(facecenteredtetragonal,fct)[9][10]���@�N�L������ĽY�����Hʹ�^�ɽ��ٵ��{�����õõ���ְl�]�������c�Kԭ���γɵĻ��W�I�ܸ��ӷ����^�ɽ���ԭ�ӣ�ʹ�䲻�������ԗl�����ܽ⣬�M�����Mһ��������˴����Ժͷ����ԡ������ꌦPtM�����g��������о��Y����������������Y���Ľ����g����������ȟo��Y���ĺϽ�������F�����õĴ����Ժͷ����ԡ�����2004�꣬Abruña[11]�о�С�M�͈������������g������늴�����ȼ��늳��еđ��ã������Ƃ���һϵ�е�Pt�������g������l�F�@Щ�������F�����K�Ͻ�ͼ������K���ߵĴ����ԡ�
����3.Pt�������g������
����3.1.PtFe�����g������
���������g��������������������Y��������F�������������ͻ��W���ܣ������ڑ�����Ҫ�����鳬���ܶȴ��Դ惦���϶������ˏV�����Pע��ShouhengSun������2000�����ˆη�ɢPtFe�{���w�����Ƃ䷽������������Ͱ������Һ�У��������û��W߀ԭ������ͪ�K(Pt(acac)2)�͟���ʻ��F(Fe(CO)5)�ķ������õ��˟o��Y����PtFe�{���w�������^500��ğ�̎�����o��Y���D׃������Y����fct-PtFe�{������[12]�������^�m�о���fcc-PtFe��fct-PtFe�{���w����0.5MH2SO4�е�ꎘO��߀ԭ�����ԣ��l�F����Y����PtFe�{���w�����F���ȟo���w�����ߵĴ����ԣ�������������Һ�У�����Y����Fe��������ֻ��3.3%�����o��Y����Fe�����ēpʧ�_��36.5%������f��������Y�������Fԭ�ӵķ�������[13]��������ߴ�Ч�ʺ��F���ٵ������ʣ���������һ��Ҫ��������ı���e�c�w�e�ȣ����^С���w���ߴ���������ȱ���e������F���ٵ�������[14]�����ߜ؟�̎���^������������{���w���ĈF���L��ʹ�����ij�ʼ�����½����ڽ������о��У������ڟ�̎�����^���У�����MgO����PtFe-Fe3O4�w�����õ���ȫ����Y����fct-PtFe�{���w�����@һ������ORR�O�������yԇ�б��F��0.958V�İ벨�λ������0.9V�ıȱ���e�Ȼ��Ժ��|���Ȼ��Էքe�_��3.16mA/cm2��0.69A/mgPt�����@���ڲ�������Y���ͼ��K����[15]��
������һ���棬���Ƃ��^���в���̼�ӱ��oҲ����ֹ�{���w���L���һ�N������XinxinDu���˲��û��W������e������Fe(CO)5�������C2H2��̼Դ���Ƃ�õ�Fe/C�����ٲ���Һ��W߀ԭ����������Fe/C����������e�K�{���w������߀ԭ�^���У������F�F�ر��������c�K�x�Ӱl���ÓQ����������γ�PtFe�w�����Ǿ�̼�Ӱ����ĽY�����ڽ����ğ�̎���^����ֱ���D���������PtFe�{���w������ƽ��ֱ���s��3.6�{�ס����õĴ�����0.1MHClO4��Һ�еİ벨�λ�_��0.89V�����^3000���λѭ�h�벨�λ�½���59mV����Pt/C�����İ벨�λ�½���140mV������f����������Ĵ������Եõ����[16]��DongYoungChung���ˈ���˲��þ۶�Ͱ�����fcc-PtFe�{���w����Ȼ���ڼs700��l����̎�����õ������fct-PtFe���������@һ�^���У��۶�Ͱ�ԭλ̼����ֱ���γɵ����s��̼�ӣ������ڼ{���w�����棬��Ч����ֹ��F���L����0.9V����ȱ���e�Ȼ��Ժ��|���Ȼ��Էքe��2.3mA/cm2��1.6A/mgPt�����ң����λѭ�h�ĉ����yԇ�У�ԓ�������F���ܺõķ����ԣ����]�����ܵ�˥�p[17]��ChanwonJung���˲���Һ��߀ԭ�ķ������Ƃ���Pt-Fe�Ͻ�{���w��������ؓ�d��̼�d�w�Ϻ���1,2ʮ���������������ͪ�F��̼Դ��������γɰ����ӣ����^һϵ�П�̎����õ������ɢ����������g������Pt3Fe/C�{���w����ԓ�������|���Ȼ����_��0.454A/mgPt������̼�ӵı��o���ã����L�r�g��ѭ�h�^���Л]�аl���w���ĈF�ۺ��F���ܳ���DFT��ՓӋ��������Fԭ������������g�Y���о��бȺϽ��и��ߵ��ܽ��λ������ȺϽ�Pt-Fe/C���õķ�����[18]��
�����S���Ƃ乤ˇ�IJ�����M��Ŀǰ���ö�Ԫ��߀ԭ�ķ������Ѳ���Ҫ�ںϽ��w��������������ܫ@�������^С�������ɢ�Ľ����g�{���w�����̘I������[19]����˸��M��Ԫ�������Ƃ�̼�d�K�F�Ͻ����ǰ��w��Ȼ���ڶ��Ԛ��w�h�����џ������o��Y���Ͻ��D��������Y�����K�F�����g��������������ô����w���ijߴ�ֲ���4nm~6nm���Ҿ���ؓ�d���d�w�ϣ�늻��W�yԇ�Y���@ʾ��������Ծ������̘IPt/C�������mȻPtFe�����g������{���w�����F���˸��ߵĴ����Ժͷ����ԣ����nj��Hȼ��늳��\���^���У��e����늳���ͣ�����λ�\�Зl���£�߀�Ǖ��������F�ܽ�������ܳ����F���x�Ӡ�B���ڣ��c�O��������߀ԭ�������a��H2O2���ɷ������ɾ��Џ������Ե����ɻ�(�����Dԇ��)���@Щ���ɻ��������|�ӽ��QĤ���w�ۺ��ʹ��l�������ֽ⣬�����|�ӽ��QĤ���ѣ�늳�ʧЧ�������Ŀǰ���H���ѽ�����ʹ�ú����F���ĺϽ������g������������߀ԭ������
����3.2.PtCo�����g�{�Y��
����һ����ԣ�����ֻ�д����w������ԭ�Ӳ��ܽ��|��늽�Һ�ͷ����늴�������Ҫ�l���ڼ{���w�����棬�w���Ȳ����F���ٲ��܅��Ӵ�����������F���������ʵ��¡������Mһ����ߴ����Ժ��K�������ʣ��о��ˆT�_�l�˾��к˚��Y���ļ{�״�����ͨ���Ե̓r���������ˣ������Kԭ�Ӟ隤�ӡ��˵ĽM�ɺ͚��ӵ���ò�����ߵ�����Ì��������Ԯa�����P��Ҫ��Ӱ푣���ՓӋ���о��A�yPt���p���ٺ˚��Y�������������^�õĴ����Ժͷ�����[20]���Ԫ������һ�N�O���c�K�γɽ����g��������Ҍ��K����������кܴ�������^�ɽ���Ԫ�ء�ͨ�^���_�{�غ˵ĽY���ͽM�ɣ����ԫ@����PtCo��������g�������˵ļ{���w��[21]��DeliWang����[22]���������Y����Pt3Co@Pt/C���������������K��(H2PtCl6·6H2O)���Ȼ��(CoCl2·6H2O)�ܽ���ˮ�У��ٌ�̼���d�w��ɢ��������Һ��ӟ����l���܄����������ĥ�^�ķ�ĩ��H2/N2��Ϛ��w�зփɂ��A�μӟᣬ��K�õ�����ĺ�-���Y�����������Y����������̎��ض��_��700��r���o��Y��������ȫ�D׃������Y���������w��������2~3��Ptԭ�ӵ����С���0.1MHClO4늽�Һ�У�ԓ�����ıȱ���e�Ȼ��Ժ��|���Ȼ��Էքe��Pt/C����3����9����YezhengCai����[23]�ö�Ԫ��߀ԭ�����ȵõ�PtCo�Ͻ�{���w�������Ȳ�ͬ��̎��ضȣ��l�F700��r�Ƃ��Pt3Co/C-700��PtCo/C-700���F����õĴ����ԣ�����f���ض���Ӱ�������Ҫ���ء�
�������]��x���������Փ��Ͷ��ָ��
�����@�ú�–���Y������������һ�N������ȥ�Ͻ���ͨ�^���W��늻��W�����ķ�ʽ�x����ȥ���Ͻ�{���w��������^�ɽ��٣��ٽ��^��̎��ʹ����Ptԭ�����ţ��γ����ܵĚ��ӽY��[24]��JunruiLi����[25]����Һ�߀ԭ�����Ƃ��CoPt�{���w�������e��̼�d�w�Ϻ��M�е�һ�Ο�̎�����õ���������Y����L10-CoPt/C�{���w�����ٌ�ԓ�w����0.1MHClO4��Һ��60�����24С�r�����x�������95%Ar��5%H2��Ϛ��w��400���̎��2С�r����K�õ�L10-CoPt@Pt/C��—���Y���������@һ�����ıȱ���e�Ȼ��Ժ��|���Ȼ��Էքe�_��8.26mA/cm2��2.26A/mgPt���քe���̘IPt/C��38����19��������ģ�Mԓ�����ڌ��H늳��еķ����ԣ������ϻ������60��ėl�����M�У����^30,000��ѭ�h��L10-CoPt@Pt/C�ĘO���������]�аl��׃�������F�˷dz��õķ����ԡ��ڌ��Hȼ��늳yԇ�У�0.9V�r�������|���Ȼ��Ԟ�0.56A/mgPt��30000��ѭ�h��ֻ��19%�Ľ��͡�ͨ�^DFT��ՓӋ�㌦�Ȳ�ͬ���������K�������ܵĽY�����������Kԭ�ӌӺ����ͬ�r����CoPt��˵Ĵ������挦��߀ԭ�������g�a��������ܱ��������C������ͨ�^�������ú���λ���Ì����������M����Ч�{�أ�ʹ�����������߀ԭ�����İl����Ӌ��Y��ͬ�r�C����Coԭ�ӌ����摪���ĸ�׃���Ï���Feԭ�ӣ��@Ҳ��L10-CoPt@Pt/C�������܃���PtFe��������Ҫԭ���@�N��PtCo�����g�������ˣ������Kԭ�Ӟ隤�ĺ˚������������ĝ������ɞ����Pt/C��ꎘO��߀ԭ������
SCISSCIAHCI